This website is intended to provide learning resources and information about VEOZAH to Canadian healthcare professionals. Please select your profession type and province, and enter your medical license number to access our VEOZAH website.
All trademarks are the property of their respective owners.
© 2025 Astellas Pharma Inc. or its affiliates.
![]()
MAT-CA-VEO-2024-00042 07/25


The efficacy of VEOZAH was evaluated in the first 12-week, randomized, placebo-controlled, double-blind portion of the SKYLIGHT 1 and 2 Phase 3 studies. After the first 12 weeks, women on placebo were then re-randomized to VEOZAH for a 40-week extension to evaluate safety for up to 52 weeks total exposure. Coprimary endpoints were mean change from baseline in moderate to severe VMS frequency and severity at Weeks 4 and 12.1
The efficacy of VEOZAH for the treatment of moderate to severe VMS due to menopause was evaluated in the first 12-week, randomized, placebo-controlled,
double-blind portion of two identical Phase 3 studies. In each of these two trials, after the first 12 weeks, women on placebo were then re-randomized to VEOZAH for a 40-week extension to evaluate safety for up to 52 weeks total exposure.1
Coprimary Endpoints1:
Mean change from baseline in moderate to severe VMS frequency and severity to Weeks 4 and 12
Participants in the Study1-3:
SKYLIGHT 1 and 2 Trial Design1-3:




REFERENCES: 1. VEOZAH Product Monograph. Markham, ON: Astellas Pharma Canada, Inc. 2. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102.
3. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT.
J Clin Endocrinol Metab. (Epub) 02-03-2023.
REFERENCES: 1. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102. 2. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. (Epub) 02-03-2023.
REFERENCES: 1. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102. 2. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. (Epub) 02-03-2023.


SD: standard deviation; SE: standard error.
* Statistically significantly superior compared to placebo at the 0.05 level with multiplicity adjustment.1
FREQUENCY: Measured as a daily mean and analyzed as weekly average (calculated as the average frequency over nonmissing days from 7 days).3,4
LS Mean: least squares mean estimated from a mixed model for repeated measures analysis of covariance.1


SD: standard deviation; SE: standard error.
* Statistically significantly superior compared to placebo at the 0.05 level with multiplicity adjustment.1
FREQUENCY: Measured as a daily mean and analyzed as weekly average (calculated as the average frequency over nonmissing days from 7 days).3,4
LS Mean: least squares mean estimated from a mixed model for repeated measures analysis of covariance.1


SD: standard deviation; SE: standard error.
† Statistically significantly superior compared to placebo at the 0.05 level with multiplicity adjustment.1
SEVERITY: Measured as a daily mean and analyzed as weekly average.3,4
LS Mean: least squares mean estimated from a mixed model for repeated measures analysis of covariance.1


SD: standard deviation; SE: standard error.
† Statistically significantly superior compared to placebo at the 0.05 level with multiplicity adjustment.1
SEVERITY: Measured as a daily mean and analyzed as weekly average.3,4
LS Mean: least squares mean estimated from a mixed model for repeated measures analysis of covariance.1


Mean change in the frequency of VMS from baseline to each visit in the extension period was an exploratory endpoint. Assessments after the 12-week placebo-controlled period were descriptive only.2,4
The duration of this study is longer than that of the efficacy data in the VEOZAH Product Monograph.


Mean change in the frequency of VMS from baseline to each visit in the extension period was an exploratory endpoint. Assessments after the 12-week placebo-controlled period were descriptive only.2,4
The duration of this study is longer than that of the efficacy data in the VEOZAH Product Monograph.


Mean change in the severity of VMS from baseline to each visit in the extension period was an exploratory endpoint. Assessments after the 12-week placebo-controlled period were descriptive only.2,4
The duration of this study is longer than that of the efficacy data in the VEOZAH Product Monograph


Mean change in the severity of VMS from baseline to each visit in the extension period was an exploratory endpoint. Assessments after the 12-week placebo-controlled period were descriptive only.2,4
The duration of this study is longer than that of the efficacy data in the VEOZAH Product Monograph.
The safety profile of VEOZAH was evaluated in Phase 3 clinical studies
TWO identically designed Phase 3 efficacy and safety studies that were randomized, placebo-controlled, double-blind for 12 weeks, followed by rerandomization of women previously receiving placebo to VEOZAH (women on VEOZAH remained on VEOZAH) for an additional 40 weeks of uncontrolled treatment.
ONE Phase 3, 52-week, randomized, placebo-controlled, double-blind study evaluating long-term safety.
The most common adverse drug reaction during the 12-week placebo-controlled period in SKYLIGHT 1 and 2 (≥3% in patients receiving VEOZAH 45 mg and greater than placebo) was liver test elevation (3.2%).1
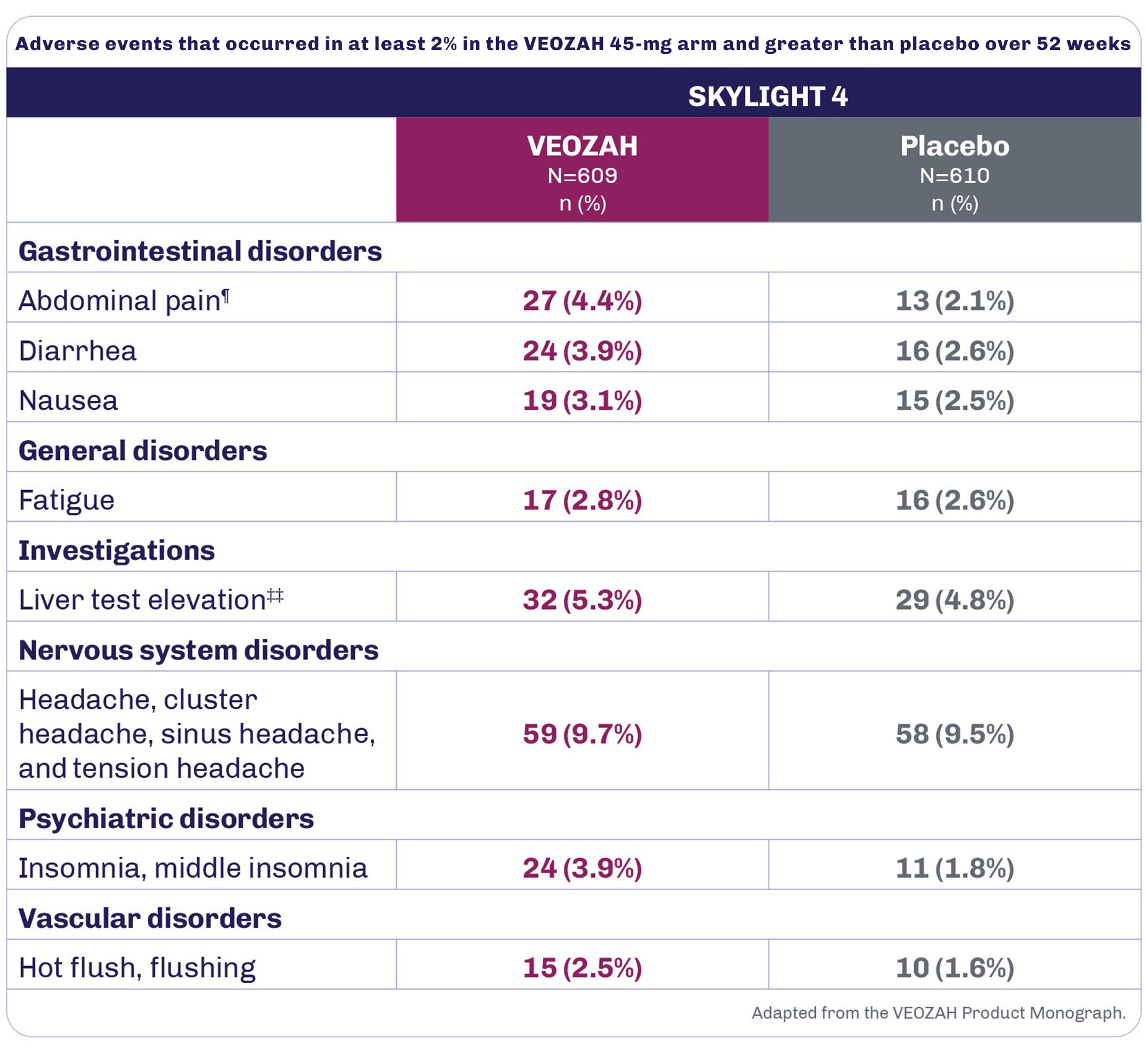
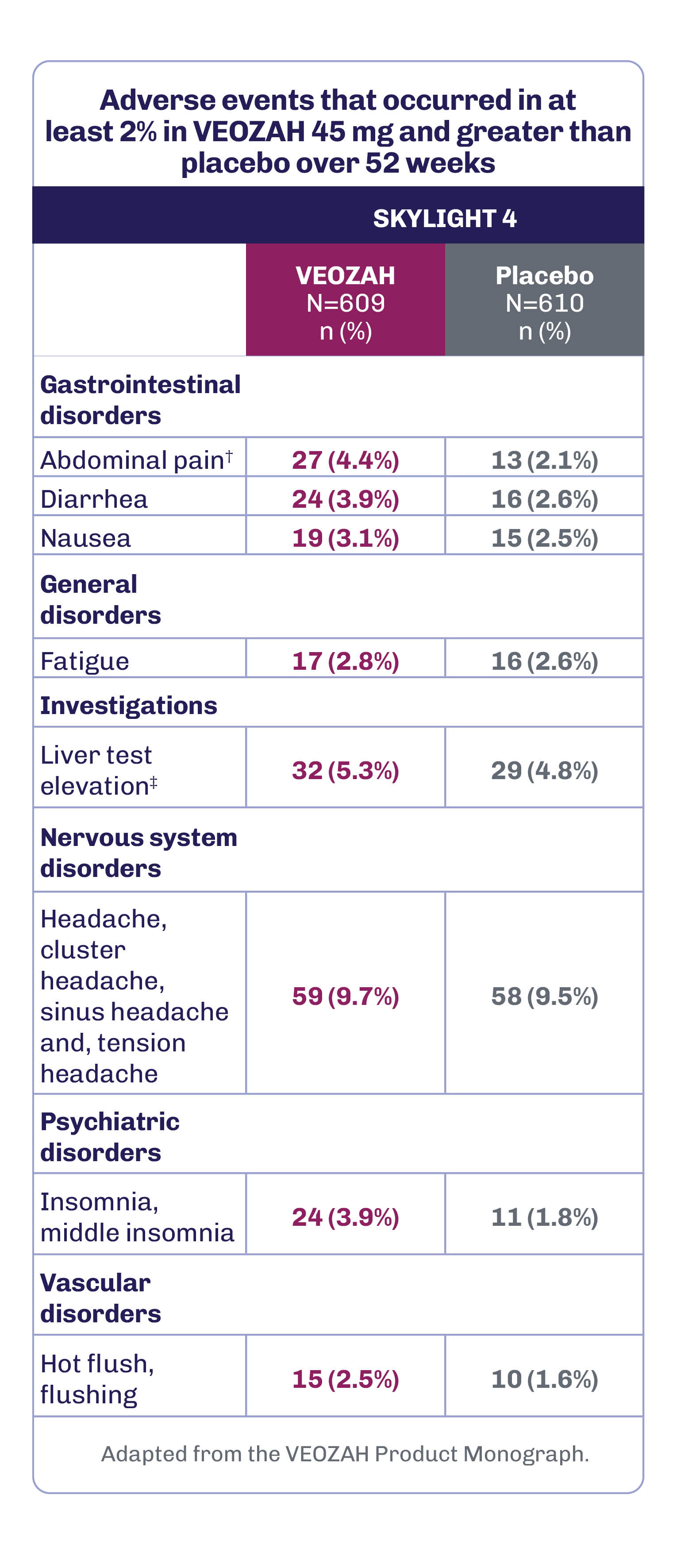
During the 52-week placebo-controlled period in SKYLIGHT 4, the most frequent adverse drug reactions (≥3% in patients receiving VEOZAH 45 mg and greater than placebo) were headache (9.7%), liver test elevation (5.3%), abdominal pain (4.4%), diarrhea (3.9%), insomnia (3.9%), and nausea (3.1%).1
A numerical imbalance was observed in the incidence of other malignancies between VEOZAH and placebo groups in the long-term safety SKYLIGHT 4. A causal relationship between VEOZAH and increased risk of malignancies has not been established.1
SKYLIGHT 4 was a Phase 3, 52-week, randomized, placebo-controlled, double-blind study evaluating long-term safety in women with VMS associated with menopause.1
Primary Endpoints2:


Demographics and Baseline Characteristics:2
The mean age of the post-menopausal women was ~55 years. Women self-identified as Caucasian (79.9%), Black or African American (17.2%), Asian (1.6%) and more than 1 race (0.9%). Ethnicity was defined as Hispanic or Latina (20.1%) or not Hispanic or Latina (79.9%). The study population included post-menopausal women with prior hormone therapy use (17.0%), with prior oophorectomy (13.5%), or with prior hysterectomy (18.6%). Body mass index was 28.4 ± 4.6. The percentage of current smokers was 19.1%.
REFERENCES: 1. VEOZAH Product Monograph. Markham, ON: Astellas Pharma Canada, Inc. 2. Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141(4):737-47.
REFERENCE: 1. Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141(4):737-47.
REFERENCE: 1. Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141(4):737-47.
*Comparative clinical significance unknown.
†SKYLIGHT 2 was a Phase 3, randomized, double-blind, placebo-controlled, parallel-group, study in which post-menopausal women were randomized to VEOZAH 45 mg (n=167) or placebo (n=167) once daily for 12 weeks. After the 12-week, double-blind treatment period, all patients received VEOZAH for a 40-week extension treatment period with women on placebo re-randomized to VEOZAH to evaluate safety for up to 52 weeks total exposure. The study included post-menopausal women who had a minimum average of 7 moderate to severe VMS per day. The coprimary efficacy endpoints were change from baseline in moderate to severe VMS frequency and severity to weeks 4 and 12. Baseline mean frequency of moderate to severe VMS per 24 hours: 11.8 for VEOZAH, 11.6 for placebo.1
‡Clinical significance is unknown.
REFERENCES: 1. VEOZAH Product Monograph. Markham, ON: Astellas Pharma Canada, Inc. 2. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102. 3. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. (Epub) 02-03-2023. 4. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study (supplementary appendix). Lancet. 2023;401(10382):1-14. 5. Data on file. Clinical efficacy summary.